It is well known that scientific developments in biologic drugs—therapies manufactured in living microorganisms, or plant or animal cells—are rapidly advancing and continue to offer great promise for patient care. Up until 2009, competition and access to these drugs was limited in the US because there was no regulatory pathway for a product similar to an existing biologic, i.e., biosimilar. But, in 2009, Congress enacted the Biologics Price Competition and Innovation Act, which called on the FDA to create such a pathway, comparable to one in place in Europe since 2005. The aim was to improve access to biosimilar drugs in the US, which were estimated to save the US healthcare system up to $44 billion over the next ten years.
Safety and Competition Concerns
For a hundred years, federal law has required that all drugs adhere to public quality standards—part of ensuring safety and protecting the public’s health. USP is the non-governmental, scientific institution charged with developing the standards to ensure the quality of medications. But language that would exempt biologics, including biosimilars, from adhering to the same public quality standards as other prescription medicines was recently added to the FDA and NIH Workforce Authorities Modernization Act (included in “Cures” legislation) by the U.S. Senate Committee on Health, Education, Labor and Pensions.
In the media, this provision has been characterized as one that would spur innovation by removing the requirement that biologics adhere to public standards. But the proposal strips biologics of the protections provided by public quality standards and unravels a critical piece of the overall safety net for these drugs. It could also hurt competition, product development and the public’s confidence in biologics and biosimilars. Without public standards, biosimilar product developers would have no common quality benchmark, which could potentially slow down the progress of new entrants into the market and impact patient access.
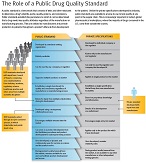
USP ensures that drug quality standards are developed through an open process with public input and are available to manufacturers and others to test a product at any point in the supply chain. In the absence of public standards, there only would be the private specifications between a manufacturer and the FDA, and they would not be publicly available. And, not having a public standard would inhibit our efforts to ensure quality across the supply chain, identify counterfeits and perform many other important public health objectives. (Click on image to view infographic on public standard vs. private specifications.)
The Critical Role of a Public Standard in Ensuring Safety, Quality and Competition
A public quality standard generally has two components: a monograph and a reference standard. USP’s monographs appear in the United States Pharmacopeia–National Formulary (USP–NF) and include tests and procedures that establish the identity, strength, quality and purity of drug products and active pharmaceutical ingredients. Reference standards are physical reference materials that are used by manufacturers to ensure that their products meet monograph requirements. Together, monographs and reference materials comprise the public standards which are the quality safety net to protect patients and ensure quality medicines.
USP’s standards are established by the USP Council of Experts—a multidisciplinary group of scientific experts from industry, academe, regulatory science and healthcare practice—with input from FDA representatives on USP Expert Committees. We also engage the 450 member organizations of the USP Convention—many of which represent patients, physicians, pharmacists and others who benefit from our public standards—in a transparent, consultative process where solid science is the decision driver. And, to supplement this robust process, USP works with the regulators from around the world to leverage their expertise to develop the best quality standards for the American public.
USP has over 65 years of expertise in developing quality standards for biologicals, starting with heparin, insulin and most recently for filgrastim. Moreover, dozens of other countries have chosen to recognize USP standards in their own country’s laws, most of which having done so in the past 20 years. Yet this proposal in Congress would exempt biologicals in the US from USP’s quality standards.
USP’s Monograph Revision Processes Allow for Continuous Updating of Standards to Reflect New Technology
USP works closely and collaboratively with FDA and both the innovator industry and biosimilars developers to evolve its standards to reflect the rapidly developing new technologies in the biologics space. To augment USP’s ongoing revision processes, the Pending and Accelerated Monograph Revision processes have been implemented within the past few years to allow USP to work quickly and proactively to update standards to reflect new technology in product specifications. One recent example is USP’s monograph for the blockbuster drug enoxaparin sodium. It has been revised several times, including through Accelerated Revision, to accommodate subsequent US market entries for this product. And, as always, we continue to engage with our broad array of scientific experts on the USP Council of Experts, the stakeholders in the USP Convention and regulators from around the world to ensure that USP monograph development is aligned with technology advances.
While our processes have evolved (and will continue to evolve) with science and the regulatory pathways established by Congress and the FDA, there is no doubt that public quality standards have been a lynchpin in protecting the quality of the US medicines supply. We need to keep them in place.
We welcome your comments and questions on this important public health issue.
Please stay tuned. We will be back in touch with you on this.
Ron
Ronald T. Piervincenzi, Ph.D. is the chief executive officer of USP.